Technology
Targeted protein degradation (TPD) utilizes the waste disposal system of the cell to selectively eliminate disease-causing proteins. As such, it is a promising alternative to existing therapeutic modalities in addressing high unmet medical needs.
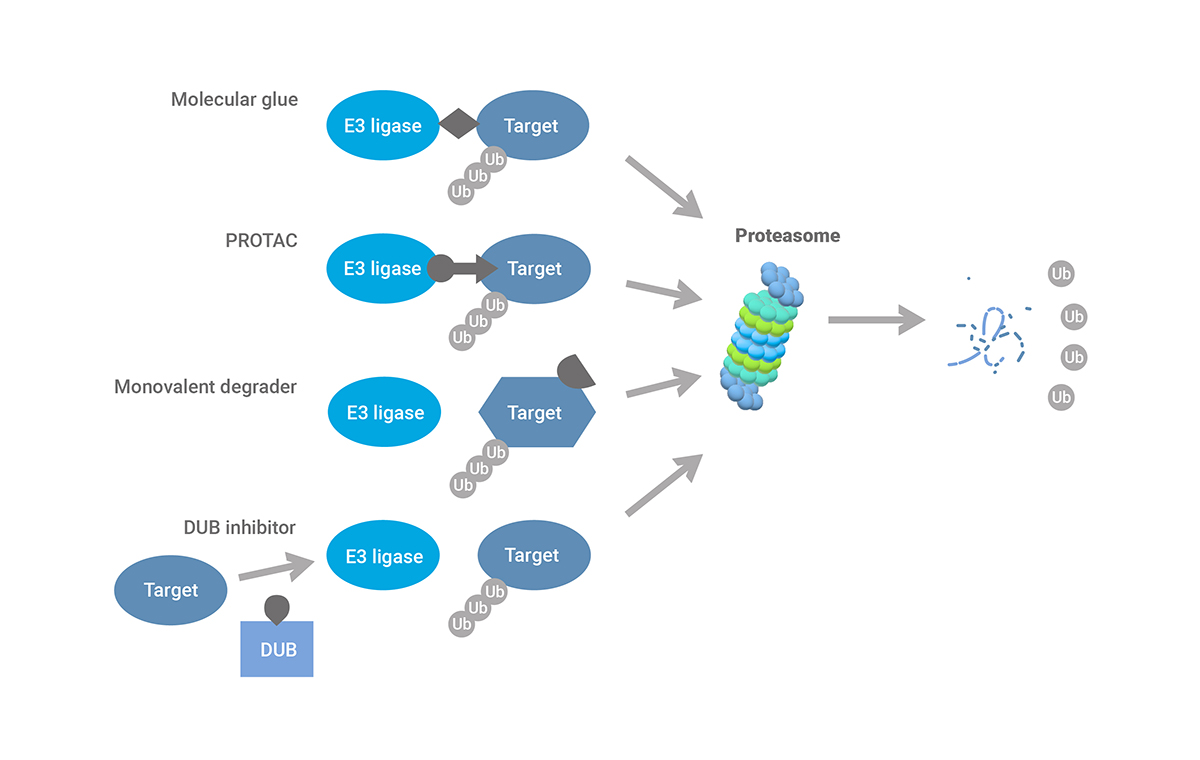
Drug discovery concepts in TPD are based on small molecules commonly referred to as ‘degraders’. Many degrader drugs redirect E3 ubiquitin ligases to non-native substrate proteins. After forming a ternary complex, the E3 ligase modifies these ‘neosubstrates’ by attaching multiple ubiquitin molecules. The proteasome recognizes the resulting poly-ubiquitin chains and degrades the neosubstrates into amino acids that are recycled for novel protein synthesis. Unlike conventional small molecule drugs, degrader molecules don’t just inhibit their targets. Instead, they act as catalytic agents that eliminate disease-causing proteins and their associated functions.
Established degrader drugs that reprogram E3 ligases come mainly in two varieties: heterobifunctional molecules also known as PROTACs (PROteolysis TArgeting Chimeras) or molecular glue compounds. PROTACs have warheads for the E3 ligase and the target protein connected by a linker. Molecular glue degraders are smaller in size and lack discernable binding units. They reshape the substrate interaction site of E3 ligases to create complementary binding sites for non-physiological targets.
Other degrader drugs, like monovalent degraders, induce depletion by triggering conformational changes in their target proteins rather than by repurposing an E3 ligase. Another strategy is to inactivate deubiquitinases that remove poly-ubiquitin conjugates. Inhibitors of these enzymes promote ubiquitination and proteasomal degradation of substrates implicated in disease. Finally, new TPD approaches go beyond the proteasome, exploiting autophagic or lysosomal degradation pathways to eliminate extracellular targets and protein aggregates.
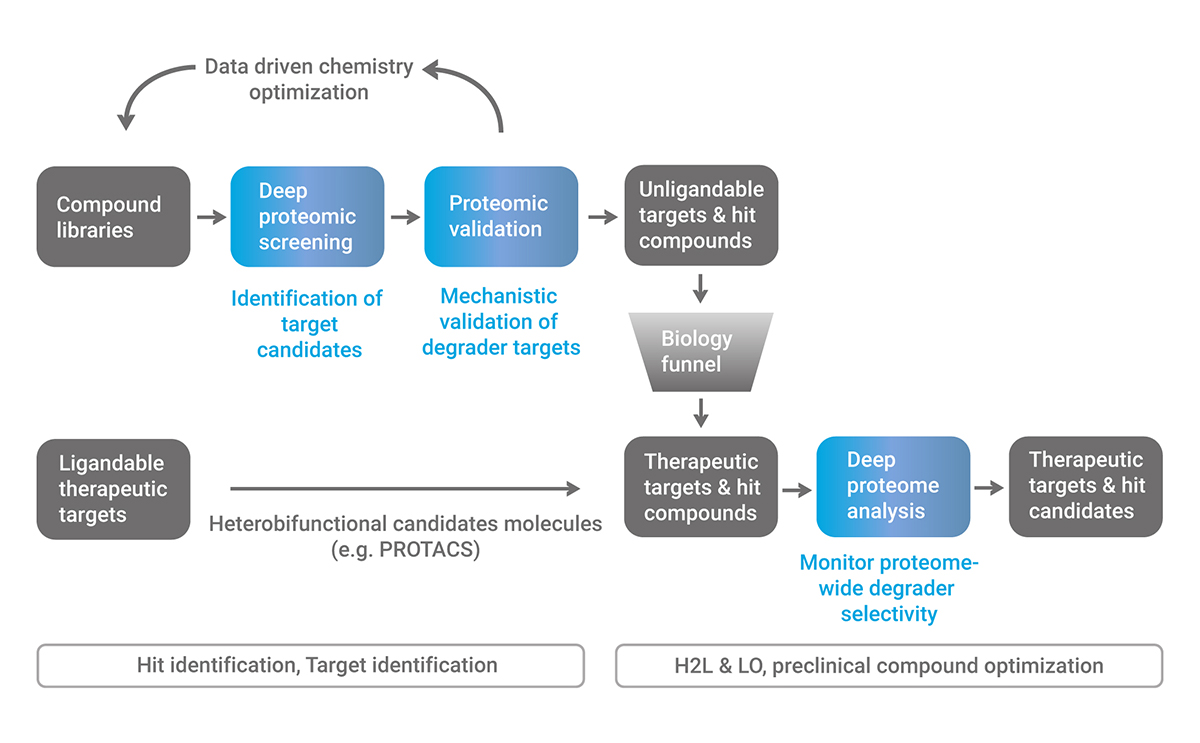
Discovery efforts in targeted protein degradation start either from degrader targets or from degrader chemistries. Both strategies benefit from deep proteomic screening to guide drug development.
The first case applies to heterobifunctional molecules like PROTACs, which are rationally designed to possess binding moieties for harmful proteins. Monitoring their selectivity against all cellular proteins provides crucial information for optimization efforts. In general, proteome-wide selectivity analysis should be performed for any degrader molecule under development for a known target.
In the second case, drug discovery originates from small molecules presumed or designed to be degraders. Testing such compounds against entire proteomes enables systematic identification of their targets. For example, molecular glue compounds, like those that bind to the E3 ligase cereblon, have been clinically validated to eliminate disease-causing proteins. With low-molecular weight, favorable drug-like properties, and the ability to promote target recognition in the absence of cavities or binding pockets, molecular glues can address proteins considered ‘undruggable‘ by conventional small molecule drugs. Their discovery, however, has so far relied on serendipitous observations that have led to only a few dozen potential neosubstrates. Thus, the true target scope of molecular glue degraders is largely unknown, despite their enormous therapeutic potential. Only a systematic, proteomics-based discovery approach that connects degrader compounds with targets in an unbiased manner can exploit the full targeting potential of molecular glues.
Cutting-edge technology that adds significant value to the drug discovery process
Our target and E3 ligase agnostic screening is suitable for characterizing all types of degrader libraries (e.g., PROTACs, molecular glues, monovalent degraders, DUB inhibitors). Putative degrader targets found in the proteomics screens can readily be validated using our unique proteomics-based validation pipeline. Besides identifying neosubstrates at unprecedented scale, the resulting data can be used for degrader library optimization and expansion, as well as for computational approaches such as AI-based predictive modeling of novel degrader drugs. The high-throughput and fast turnaround capabilities of our platform also support proteome-wide selectivity profiling in drug optimization cycles.
We believe that only proteomics using mass spectrometry at unprecedented scale, depth, and quality can reveal the true target scope of TPD. Proteomics will identify novel, previously undruggable targets and advance degrader drugs to develop treatments for life-threatening diseases. Our unique, integrated proteomics platform transforms today’s challenges in targeted protein degradation into tomorrow’s opportunities.
 DEEP
DEEPPROTEOMIC
SCREENING
 FULLY AUTO-
FULLY AUTO-MATED AND
SCALABLE
WORKFLOWS
 HIGH-QUALITY
HIGH-QUALITYDATA
DELIVERY
 EXPERT DATA
EXPERT DATAHANDLING AND
ANALYSIS
 PROTEOMIC
PROTEOMICVALIDATION
OF DEGRADER
TARGETS
 SYSTEMATIC
SYSTEMATICMAPPING OF
NOVEL
TARGET SPACE
© NEOsphere Biotechnologies GmbH
All rights reserved
Service links
Imprint
Data Policy
Company Brochure
Contact
NEOsphere Biotechnologies GmbH
Fraunhoferstrasse 1
82152 Planegg
Germany
Phone: +49 89 4378 020 0
Mail:
